
Christopher Cheng, Ph.D., and Valerie Merkle, Ph.D.
This is the third of five short articles on the importance of cardiovascular device biomechanical compatibility. These articles discuss: 1) why biomechanical compatibility is important, 2) its role in the device development process, 3) fatigue evaluation for initiating clinical trials, 4) fatigue evaluation for market approval, and 5) the future of biomechanical compatibility assessment.
It's All Fun and Games Until
During the medical device development process, the stakes rise with every stage, transitioning from concept discovery, to design iteration, preclinical testing, clinical testing, and finally, market launch. These stages often cross and blend together, but there is no denying that a major nail-biting moment is moving into initial clinical evaluation. This is the first time human lives may be at stake. Sufficient information, including evidence related to device durability, must be generated to support that the device is unlikely to cause undue morbidity or mortality. This does not mean that all device fatigue risk must be driven out – that is an impossible task. However, enough fodder must be gathered to justify the leap to subjecting a small number of patients to unknown risks in the effort to help the larger patient population.
Clinical Indication and Clinical Study Phase
The burden of proof related to mechanical durability to initiate a clinical study is highly dependent on the clinical indication. Is the device part of an acute treatment or something that needs to last a lifetime? Are the patients elderly with limited mobility and a short life expectancy, or are they children with treatable congenital heart defects who may live 90 years and will someday do an Ironman Triathlon? Are the patients seeking a last resort after all other options have been exhausted, or are there reasonable treatment alternatives available? The scope of durability evaluation depends on these factors of dwell time, loading magnitudes, and the safety of other treatment options. Another major factor in determining the burden of proof is the phase of the clinical study.
Appropriate Evaluation at the Appropriate Stage
Often, the primary goals of a clinical study are to collect safety and effectiveness information of a device, however, critical data on design weaknesses can also be gleaned. The earlier in the device development process, that is, the more a device has yet to be optimized, the leaner the durability assessment often is. While fatigue evaluation must still de-risk the device enough to warrant exposure to patients, there is no need to complete market approval-levels of evaluation in early studies because the clinical experience may yield information that leads to substantial design changes. The amount of investment that should be dedicated to these appropriately supportive, but as yet inexhaustive, fatigue evaluations are also dependent on knowledge of the device category at large. A device that is an iterative improvement on existing devices may be able to leverage existing durability data while a truly breakthrough device may require more evaluation before clinical exposure. Regarding investment level, manufacturers must also consider the amount of development risk (i.e. potential time and cost of changing design) and regulatory risk (e.g. need for additional data and regulatory reviews) to take at each stage.
Clinical Data is Part of Biomechanical Evaluation
A concept that bears highlighting is that a clinical study can itself generate biomechanical boundary conditions that lay the groundwork for realistic durability evaluation. This means that device durability evaluation should not be relegated as an inconvenient input prior to clinical studies, but clinical studies may in fact generate critical outputs that aid in durability assessment. Thus, there are prudent opportunities to leverage costly clinical studies to generate necessary engineering data with low incremental investment. While device-specific in vivo data may not be necessary for pure force-controlled or displacement-controlled loading conditions, devices that exist in complex environments with mixed loading conditions can benefit from dedicated clinical investigation. For example, while an isolated, short arterial stent may be fatigued predominantly by radial deformation from blood pressure pulsatility, a bridging stent in complex endovascular aortic repair is subject to bending forces as a result of complex interactions between the bridging stent, main endograft, and diaphragmatic and visceral organ translation due to respiration (Figure).
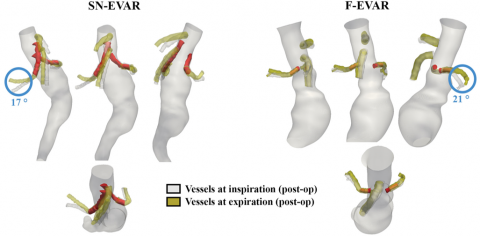
Figure. Respiratory-induced deformation of the renovisceral arteries for examples of snorkel- (SN-EVAR) and fenestrated- (F-EVAR) endovascular aortic repair. Vessels at inspiration (gray) and expiration (yellow) are superimposed with the bridging stents (red). Respiratory-induced end-stent angle changes are highlighted with blue circles. Adapted from Ullery et al., J Vasc Surg 2015, 61(4), 875-884, Figure 3.
Teamwork
Device manufacturers can work hand in hand with their clinical investigators and clinical research organization (CRO) to generate the biomechanical data needed to optimize devices and device evaluation. Working with the FDA to come up with a plan for the right testing at the right time, and protecting patient safety while collecting informative biomechanical data is critical to reduce both financial and patient risks. Additionally, it is imperative to communicate with the FDA through a well-written pre-submission and incorporate. This will improve the likelihood that the costly and time-consuming durability evaluations to support regulatory submissions will meet the expectations of the FDA reviewers. Finally, remember that the ultimate goal of timely and robust fatigue evaluation is to improve patient care.
About the Authors
Dr. Christopher Cheng has 20+ years of experience in academic research and the medical device industry, spanning hemodynamics, vascular motion, device design, manufacturing, preclinical testing, clinical trials, and marketing. He is considered the preeminent expert in vascular motion, having over 100 publications and edited Handbook of Vascular Motion (PROSE Book Award Nominee, https://www.elsevier.com/books/handbook-of-vascular-motion/cheng/978-0-1...), the first and only book dedicated to how blood vessels move. Dr. Cheng runs the Global Science & Technology – Medical Division (gst.com/med), the first dedicated organization to help medical device companies holistically evaluate and improve biomechanical compatibility of medical implants. He is also an Adjunct Professor in the Division of Vascular Surgery at Stanford, Director of the Vascular Intervention Biomechanics & Engineering (VIBE) lab (vibelab.stanford.edu), and Director of the Cardiovascular Implant Durability Conference (cvidconference.org). Previously, Dr. Cheng was co-founder and CEO of Kōli, Inc., an early-stage medical device company developing a catheter-based solution for gallstone disease. Dr. Cheng studied BME and EE at Duke University, earned his Master's and Ph.D. in Biomechanics at Stanford University, and currently serves as a board member of the Duke University Pratt School of Engineering.
Dr. Valerie Merkle is the Associate Director of Regulatory Strategy at Syntactx. With the Syntactx Team, she provides expert assistance to clients seeking regulatory approvals and product adoption worldwide. Prior to joining Syntactx, Dr. Merkle was a leader in the U.S. Food and Drug Administration (FDA) in the Center for Devices and Radiological Health (CDRH) vascular and endovascular surgical devices team. In her role, she managed and reviewed over 650 complex regulatory submissions, including pre-submissions, investigational device exemptions, as well as 510(k) and PMA marketing submissions. Her FDA experience included managing challenging submissions for first-of-a-kind devices and those with unique benefit/risk profiles, providing her the opportunity to serve as the FDA lead to a meeting of the Circulatory System Devices Panel. Her additional expertise and outreach include standards work, cardiovascular materials research, and FDA/external research collaborations. Dr. Merkle is a Steering Committee Member for the Greenberg Stent Summit, a unique conference that brings together representatives from industry, clinical practice, and FDA to discuss current issues in endovascular interventional therapy. Dr. Merkle holds a Bachelor of Science in Chemical Engineering from Bucknell University, a Ph.D. in Biomedical Engineering from the University of Arizona, and is an Innovation Fellow of the Fogarty Institute. She has co-authored numerous peer-reviewed manuscripts and has presented at multiple academic, scientific, and technical conferences.
