
Christopher Cheng, Ph.D., and Valerie Merkle, Ph.D.
This is the fourth of five short articles on the importance of cardiovascular device biomechanical compatibility. These articles discuss: 1) why biomechanical compatibility is important, 2) the role of biomechanical compatibility in the device development process, 3) fatigue evaluation for initiating clinical studies, 4) fatigue evaluation for market approval, and 5) the future of biomechanical compatibility assessment.
Prepare to Enter the Wild
The leap from preclinical to clinical evaluation is a critical one, exposing patients to potential morbidity and mortality. However, the transition from clinical study to market release is equally serious, with a much larger risk profile. The risks are not limited merely to the increase in the number of patients that may be treated by the device. Clinical studies typically operate under tight control, whereas commercialization releases a product into relatively unrestricted use. While instructions for use (IFU) are meticulously written to guide physicians on proper patient selection, treatment, and care, it is not uncommon for the device to be used outside of the IFU as part of the practice of the medicine. Furthermore, while insights into device durability, including computational, benchtop, and early clinical data, are captured during product evaluation to support regulatory approval, real-time in vivo implantation of greater than 1-3 years is often realized for the first time after market release.
Durability Prediction
Since real-time in vivo evaluation of the maximum dwell time of a device in a patient could exceed 10 or even 20 years, it is not reasonable to expect real-time fatigue evaluation prior to market approval. If this were required, countless patients could be precluded from safe and effective therapies, with this risk greatly outweighing the risk of treatment. This is precisely why non-clinical fatigue evaluation must be performed in a robust and comprehensive manner. Since fatigue is known to be a logarithmic phenomenon, that is, the risk of fracture increases less as the number of fatigue cycles increase, devices may not need to be tested out to the maximum theoretical life span of the patient. Thus, regulatory bodies typically recommend aortic and peripheral vascular devices be evaluated to 10 years of equivalent life, and heart valve devices to 15 equivalent years due to their more critical and dynamic nature (Figure). The precision of the of non-clinical fatigue evaluation may also be influenced by: a) the amount of information available to characterize the in vivo environment, b) expected patient population, and c) the level of understanding of the device interactions with the cardiovascular system.
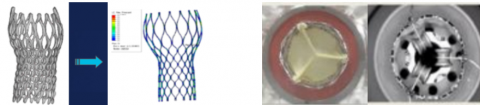
Figure. Examples of percutaneous heart valve fatigue evaluation. Left: Fatigue evaluation using in vivo deformation analysis from medical imaging and computational simulation (adapted from https://slideplayer.com/slide/4236293/, Slide 10). Right: Benchtop durability evaluation of accelerated wear and metallic frame fatigue (adapted from Cribier, Global Cardiology Science & Practice 2016: 32 http://dx.doi.org/10.21542/gcsp.2016.32, Figure 5).
Many Paths to the Summit
Each round of regulatory review consumes valuable time before commercialization and revenue generation. Providing iterative levels of mechanical durability data through a series of regulatory communications may involve a longer review process. Conversely, providing thorough durability evidence early may reduce the review timeline, but is likely to require more upfront investment in generating robust loading conditions, computational simulations, and benchtop testing. Investment in durability evaluation must be balanced with the other submission components; however, the cost of durability assessment is usually dwarfed by the costs of appraising clinical safety and effectiveness. The paths to the end goal are numerous and highly variable because fatigue assessment guidance is limited, and loading conditions, methodology, and acceptance criteria are all up to the sponsor to define and justify. Sometimes, rationale can be used to reduce the burden for test data, but only when accompanied by clear arguments that demonstrate deep understanding of fatigue mechanisms. Working with regulatory agencies from the outset, thereby minimizing incorrect assumptions, will decrease the risk of falling short or wasting resources on superfluous analysis. Importantly, durability evaluation plan must be carefully considered not merely for the purpose of satisfying regulatory agencies, but for truly addressing the clinical situation.
Life After Market Approval
A post-market study may be required by a regulatory body to evaluate longer-term clinical performance, initiated by the manufacturer to study real-world use (e.g. use of a registry) and/or performed by independent investigators. These clinical studies, if deliberately designed, may potentially provide insight into the performance of the device not gleaned from the shorter-term feasibility and pivotal studies where there was strict adherence to study protocols. Further, these post-market studies can be important for informing future durability assessments if information is obtained in the clinical study with respect to critical boundary conditions. Quality durability data can also have marketing benefit by way of presentations and peer-reviewed publications.
Regulatory Strategy is Key
Efficiency through the regulatory process can be obtained by a well-defined regulatory strategy. This begins with defining the end goals for the device early in the product development process, for example, including a broad indications for use statement and/or multiple procedural options for use of the device. The clinical and engineering data needed to support the desired labeling, including the data to support any potential interim milestones, should be outlined in the regulatory plan. For example, presenting a prospectively-defined and well-justified plan to the FDA through pre-submission interactions provides a manufacturer opportunities to collaborate with the Agency on an appropriate path forward. It is important to remember that the ultimate goal of an efficient regulatory strategy is to provide patients with timely access to safe and effective medical devices.
About the Authors
Dr. Christopher Cheng has 20+ years of experience in academic research and the medical device industry, spanning hemodynamics, vascular motion, device design, manufacturing, preclinical testing, clinical trials, and marketing. He is considered the preeminent expert in vascular motion, having over 100 publications and edited Handbook of Vascular Motion (PROSE Book Award Nominee, https://www.elsevier.com/books/handbook-of-vascular-motion/cheng/978-0-1...), the first and only book dedicated to how blood vessels move. Dr. Cheng runs the Global Science & Technology – Medical Division (gst.com/med), the first dedicated organization to help medical device companies holistically evaluate and improve biomechanical compatibility of medical implants. He is also an Adjunct Professor in the Division of Vascular Surgery at Stanford, Director of the Vascular Intervention Biomechanics & Engineering (VIBE) lab (vibelab.stanford.edu), and Director of the Cardiovascular Implant Durability Conference (cvidconference.org). Previously, Dr. Cheng was co-founder and CEO of Kōli, Inc., an early-stage medical device company developing a catheter-based solution for gallstone disease. Dr. Cheng studied BME and EE at Duke University, earned his Master’s and Ph.D. in Biomechanics at Stanford University, and currently serves as a board member of the Duke University Pratt School of Engineering. Email: winecpc@gmail.com
Dr. Valerie Merkle is the Associate Director of Regulatory Strategy at Syntactx. With the Syntactx Team, she provides expert assistance to clients seeking regulatory approvals and product adoption worldwide. Prior to joining Syntactx, Dr. Merkle was a leader in the U.S. Food and Drug Administration (FDA) in the Center for Devices and Radiological Health (CDRH) vascular and endovascular devices team. In her role, she managed and reviewed over 650 complex regulatory submissions, including pre-submissions, investigational device exemptions, as well as 510(k) and PMA marketing submissions. Her FDA experience included managing challenging submissions for first-of-a-kind devices and those with unique benefit/risk profiles, providing her the opportunity to serve as the FDA lead to a meeting of the Circulatory System Devices Panel. Her additional expertise and outreach include standards work, cardiovascular materials research, and FDA/external research collaborations. Dr. Merkle is a Steering Committee Member for the Greenberg Stent Summit, a unique conference that brings together representatives from industry, clinical practice, and FDA to discuss current issues in endovascular interventional therapy. Dr. Merkle holds a Bachelor of Science in Chemical Engineering from Bucknell University, a Ph.D. in Biomedical Engineering from the University of Arizona, and is an Innovation Fellow of the Fogarty Institute. She has co-authored numerous peer-reviewed manuscripts and has presented at multiple academic, scientific, and technical conferences.
