
Christopher Cheng, Ph.D., and Valerie Merkle, Ph.D.
This is the second of five short articles on the importance of cardiovascular device biomechanical compatibility. These articles discuss: 1) why biomechanical compatibility is important, 2) its role in the device development process, 3) fatigue evaluation for initiating clinical trials, 4) fatigue evaluation for market approval, and 5) the future of biomechanical compatibility assessment.
Starting Too Late
Let’s start with a hypothetical. A company spends three years building a team, iterating designs, screening materials, constructing a supply chain, conducting preliminary preclinical and functional evaluations, and building the bones of a design history file for an improved version of an existing medical device. Now it is time to freeze the design and launch costly good laboratory practice (GLP) evaluations necessary for getting approval for a pivotal clinical study. This includes biocompatibility, acute and/or chronic animal safety studies, simulated use, mechanical durability, package integrity, and sterility testing, among other things. Then, the company spends the next two years completing these activities and obtaining approval of an investigational device exemption (IDE), only to learn that the existing marketed device causes intimal hyperplasia and occlusion due to blood vessel kinking. While this company spent millions of dollars going through design input, design output, and design verification phases, it turns out they had not adequately defined the design inputs in the first phase. By the way, about the hypothetical assumption of only spending five years to get an IDE for a Class III permanent implant … it’s often far longer than that.
Biomechanics as a Design Input
Biomechanical compatibility and durability is often an afterthought to medical device design – in fact it should to be one of the starting points. As the first article of this series described, a device needs to perform its primary function while surviving and accommodating its environment. For example, a iliofemoral venous stent not only needs to maintain luminal area in order to promote adequate venous return, it also needs to survive radial pulsatility, resist diametric compression from the pubis bone, and bend with hip flexion without causing trauma at the end of the stent (Figure). A failure to account for these biomechanical factors during the design input phase could cause serious, and costly, delays during development.
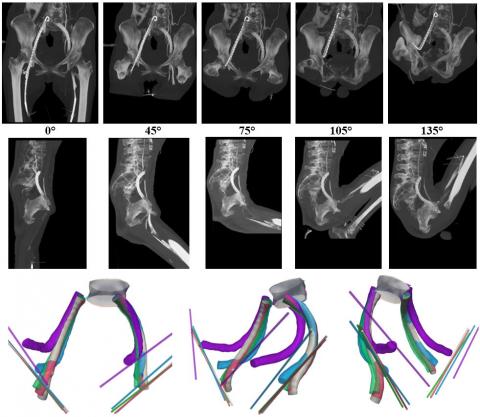
Figure. Lateral images of iliofemoral venous stents with 0, 45, 75, 105, and 135 degrees of hip flexion (top row), and 3D models of stents, L5 vertebral body, and inguinal ligament paths corresponding to 0 (gray), 45 (red), 75 (green), 105 (blue), and 135 (purple) degrees of hip flexion (bottom row). From Cheng et al., J Vasc Surg Venous Lymphat Disord 2020, doi:10.1016/j.jvsv.2020.01.022>
Leverage Existing Devices and Obligatory Development Activities
Much of determining design inputs with respect to biomechanical accommodation may be gleaned from data of existing devices. For example, let's say you are developing the first stent with an inferior vena cava (IVC) indication in the United States (none currently exist). Even without any approved predicate devices, stents and IVC filters have been implanted into the IVC for decades, and can inform how breathing and Valsalva maneuvers may deform the new device. The deformation of an existing device can be tracked on medical images, essentially playing the role of a strain gauge, and used to predict deformations of the new device by comparing the mechanical properties between the two. Furthermore, obligatory development activities to test safety and function, such as animal and cadaver studies, can often be leveraged to investigate biomechanics with some protocol tweaks.
Can Be Small Incremental Cost and Effort
Early studies to understand biomechanics do not need to be expensive. For developing loading conditions, pre-GLP and pre-clinical trial studies cost a fraction of the later, more-structured activities that will form the bulk of the eventual regulatory submission. For computational simulation and fatigue benchtop testing, shorter, less-structured, and cheaper studies will do the trick early in the development cycle, especially before design freeze. From the perspective of the entire development path, these scoping activities will only add small incremental costs and effort, and dramatically reduce development risk.
Penny Wise and Pound Foolish
Effort and costs increase with each stage of medical device development, with big jumps at design freeze, clinical study, and market rollout. This means that delays due to unexpected development activities get more costly the later they occur. A well-designed device evaluation strategy reduces costly errors during verification and validation testing by identifying the right testing to do at the right time. The strategy ideally takes into consideration relevant device attributes, potential failure modes, clinical effects of failure, and clinical mitigation strategies. When executed correctly, it will provide valuable insights into device safety and performance, with appropriate levels of testing to support each milestone (e.g., early feasibility study, pivotal study, marketing approval/clearance) beginning early in the product development process and iterating as new information is obtained. To avoid costly errors, a device evaluation strategy should not be based on the minimum regulatory requirements to support each milestone, but rather on the testing needed to further the understanding of the device and its performance throughout product development.
About the Authors
Dr. Christopher Cheng has 20+ years of experience in academic research and the medical device industry, spanning hemodynamics, vascular motion, device design, manufacturing, preclinical testing, clinical trials, and marketing. He is considered the preeminent expert in vascular motion, having over 100 publications and edited Handbook of Vascular Motion (PROSE Book Award Nominee, https://www.elsevier.com/books/handbook-of-vascular-motion/cheng/978-0-1...), the first and only book dedicated to how blood vessels move. Dr. Cheng runs the Global Science & Technology – Medical Division (gst.com/med), the first dedicated organization to help medical device companies holistically evaluate and improve biomechanical compatibility of medical implants. He is also an Adjunct Professor in the Division of Vascular Surgery at Stanford, Director of the Vascular Intervention Biomechanics & Engineering (VIBE) lab (vibelab.stanford.edu), and Director of the Cardiovascular Implant Durability Conference (cvidconference.org). Previously, Dr. Cheng was co-founder and CEO of Kōli, Inc., an early-stage medical device company developing a catheter-based solution for gallstone disease. Dr. Cheng studied BME and EE at Duke University, earned his Master’s and Ph.D. in Biomechanics at Stanford University, and currently serves as a board member of the Duke University Pratt School of Engineering. Email: winecpc@gmail.com
Dr. Valerie Merkle is the Associate Director of Regulatory Strategy at Syntactx. With the Syntactx Team, she provides expert assistance to clients seeking regulatory approvals and product adoption worldwide. Prior to joining Syntactx, Dr. Merkle was a leader in the U.S. Food and Drug Administration (FDA) in the Center for Devices and Radiological Health (CDRH) vascular and endovascular devices team. In her role, she managed and reviewed over 650 complex regulatory submissions, including pre-submissions, investigational device exemptions, as well as 510(k) and PMA marketing submissions. Her FDA experience included managing challenging submissions for first-of-a-kind devices and those with unique benefit/risk profiles, providing her the opportunity to serve as the FDA lead to a meeting of the Circulatory System Devices Panel. Her additional expertise and outreach include standards work, cardiovascular materials research, and FDA/external research collaborations. Dr. Merkle is a Steering Committee Member for the Greenberg Stent Summit, a unique conference that brings together representatives from industry, clinical practice, and FDA to discuss current issues in endovascular interventional therapy. Dr. Merkle holds a Bachelor of Science in Chemical Engineering from Bucknell University, a Ph.D. in Biomedical Engineering from the University of Arizona, and is an Innovation Fellow of the Fogarty Institute. She has co-authored numerous peer-reviewed manuscripts and has presented at multiple academic, scientific, and technical conferences.
